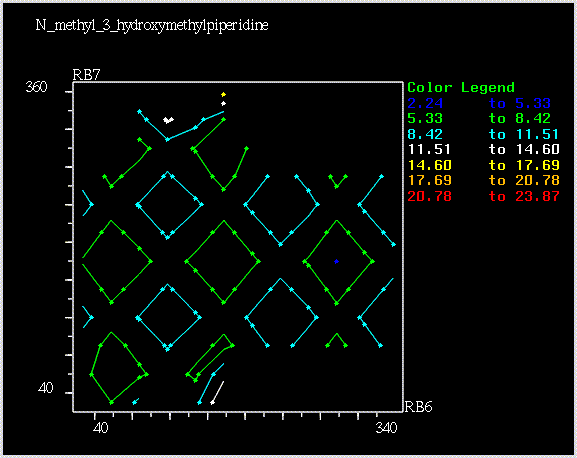
Conformational Energy Sampling Using SYBYL
The basic steps for performing energy sampling involve structure input (an energy minimized conformation or a crystal structure) and the specification of rotatable bonds and grid parameters (start, increment, stop). Conformational energy sampling is performed using the "Search" command ("Systematic Search... sub-command) of the "Compute" menu. Other utilities of the Search command ("Grid Search..." and "Random Search...") are not included in this discussion.
Searching Acyclic Bonds
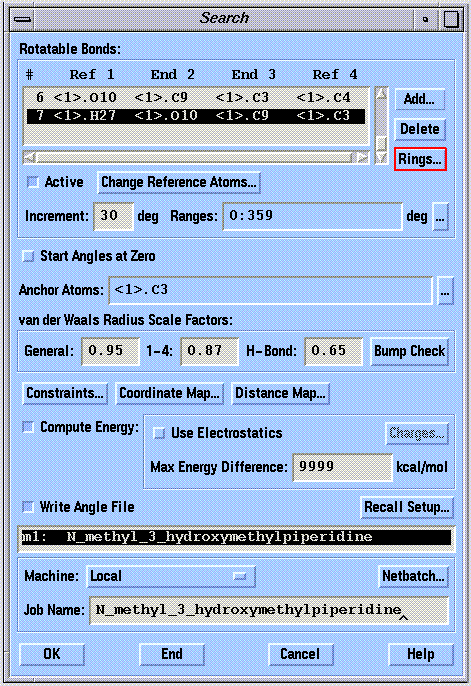
Acyclic bond rotations are input by clicking on the "Add..." button in the "Search" window:

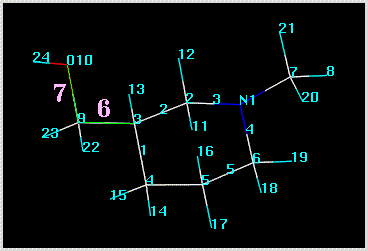
Acyclic bond rotations are specified in the form of dihedral angles. You pick the central bonds by clicking on atoms (e.g. 3,9 and 9,10), and SYBYL automatically selects the reference atoms (e.g. 4,10 and 3,24). The picked bonds are colored green:
Searching Cyclic Bonds
Cyclic bond rotations are input by clicking on the "Rings..." button in SYBYL's Search window:
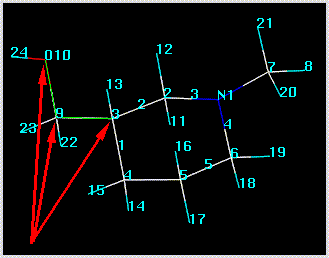
Rotatable ring bonds are automatically defined based on your selection of a ring closure bond:
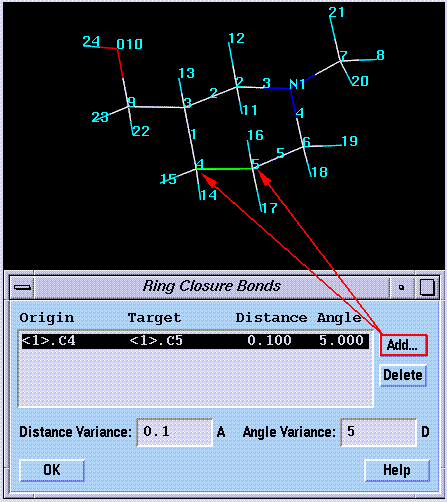
The ring closure bond is "broken" during the search. Conformations are only considered valid if the ring closure bond can be sealed with a correct valence geometry plus or minus some tolerance. The angle and distance tolerances can be increased if a search does not find a reasonable number of conformations.
Grid Setup
A systematic search should always be performed on a fully energy minimized (see gradient search energy minimization) or experimentally determined starting structure. The starting dihedral angle values of each rotatable bond can be taken from the initial structure or they can be zeroed (i.e. set to cis). The initial dihedral angle values define the reference conformation to which all generated conformations are referenced (bond rotations are relative to the initial conformation). It is usually best not to zero the dihedral angles, but to archive the initial structure for use with future search calculations.

The rotation increment and range (in degrees) must be specified for each bond included in the search. Click on the bond (in the "Rotatable Bonds" box) whose grid values you wish to change. The increment can be changed by typing a value into the "Increment" box. The rotation increment for cyclic bonds should be small (5-10 degrees) in order to find a sufficient number of ring conformations. The range can be changed by clicking on the "..." button adjacent to the "Ranges" box.
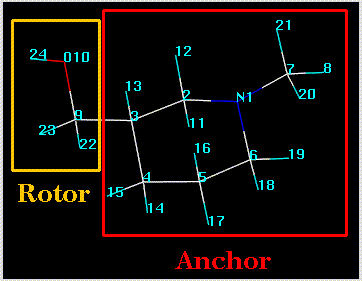
The anchor atom defines the side of a rotatable bond that remains stationary during a search. This is primarily used to maintain a meaningful spatial orientation of a structure (usually an orientation associated with superposition on another structure). The anchor atom can be changed by clicking on the "..." button adjacent to the "Anchor Atoms" box. This concept is shown in the following figure, in which atom 3 has been defined as the anchor:
Energetics Considerations and Filtering
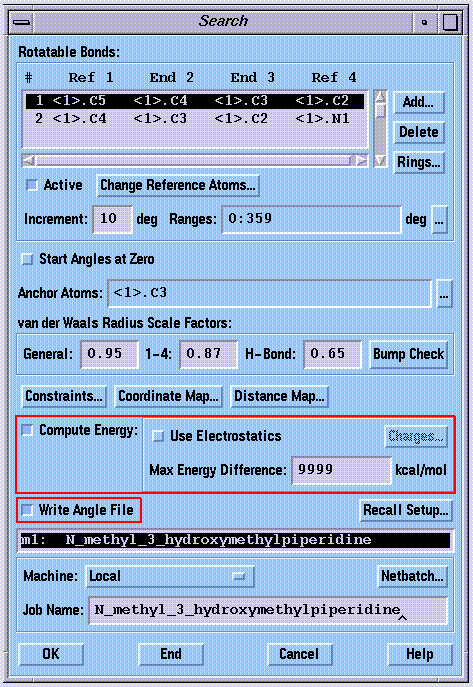
SYBYL computes and records molecular mechanics energies for each conformation sampled if the "Compute Energy" button is selected. Although the use of electrostatic energy is not recommended by the SYBYL authors, it should nevertheless be included by selecting the "Use Electrostatics" button. The partial atomic charges should be assigned using the Gasteiger-Marsili method. An absolute energy cutoff can be applied by filling in the "Max Energy Difference" box. This box is often 0.0 by default, and if left that way, will result in the loss of all conformational data from the search. If you do not want to apply a cutoff filter, enter "9999" in the box. SYBYL will not record any conformational data unless the "Write Angle File" button is selected before a search.
The primary filtering performed by SYBYL (e.g. tree pruning) is based on a check of interatomic distances between non-bonded atoms. Conformations are considered to be high in energy if any non-bonded contact distances are less than their van der Waals contact distance. The molecular mechanics energy is not used for this purpose because the computational requirements are great compared to the bump check approach.
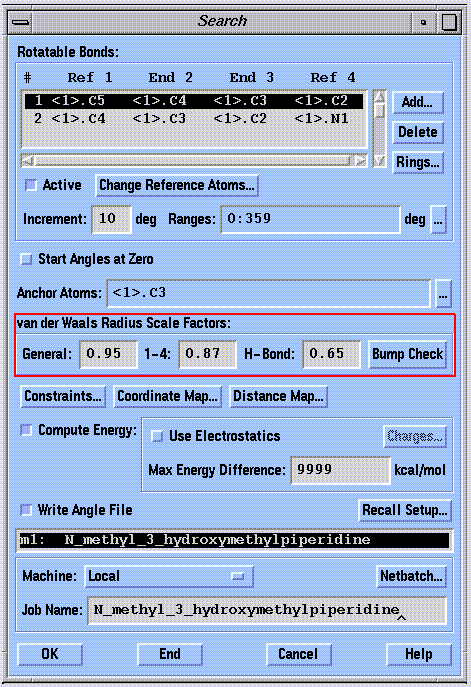
The valence geometry (bond angles and bond lengths) is held fixed during a conformational search. This approach reduces the number of degrees of freedom in the problem, but can overestimate bad contacts that could be relieved by slight flexing in a bond angle or length. SYBYL compensates for this limitation by using reduced van der Waals radii in the bump check calculation, as shown in the following diagram:

The van der Waals radii are scaled according to the constants shown in the "van der Waals Radius Scale Factors" boxes. Reducing the scaling factors decreases the stringency of all bump checks.
Performing the Search Calculations
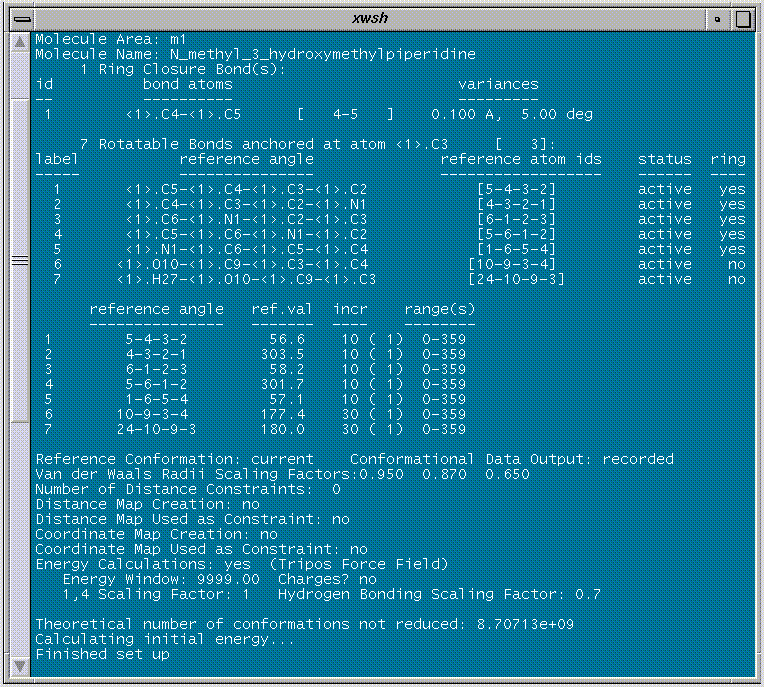
The search is performed by clicking on the "OK" button (note that the calculations are performed in batch mode). The input is summarized in the SYBYL text window:
Analysis of Conformational Data
When the calculations have completed, the number of conformations stored in the ".ang" file can be determined from the ".sta" file using the Unix "more" command. If this number is extremely large (many thousand), post-filtering of the ".ang" file based on energy should be considered (use the "Filter Search Files..." command of the "Analyze" menu). If the ".ang" file contains a reasonable number of conformations, it can be directly read using the "Systematic Search..." command of the "Analyze" menu:
Analysis of conformational data typically consists of graphical examination of the structures, sorting by energy to group the lowest energy conformations (keep in mind that these are not minima), and plotting energy vs. rotational degrees of freedom.
These tasks are greatly facilitated by SYBYL's "Molecular Spreadsheet," which is used to store the dihedral angle values and energies of all computed conformations:
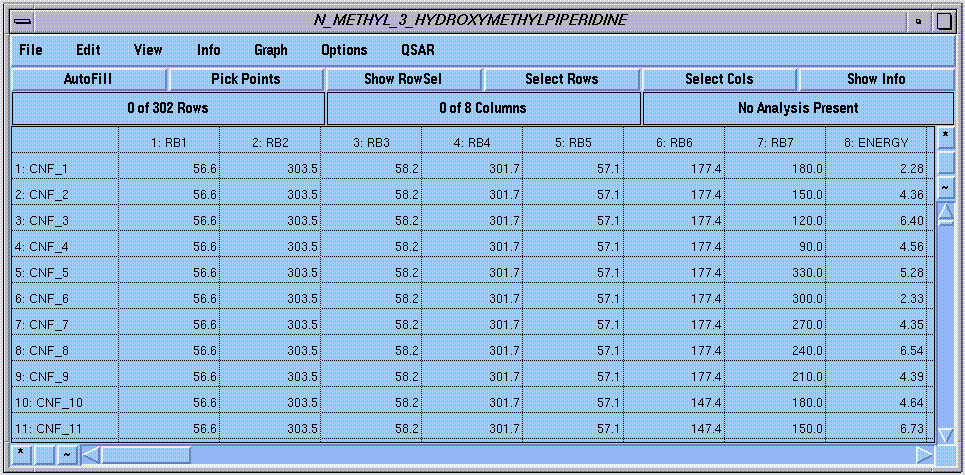
The spreadsheet commands are located in the menu bar of the spreadsheet window. These commands operate on rows and columns that you select by clicking on a specific row or column of the spreadsheet, or using the "*" buttons to select all rows or columns. The selected cells are highlighted:
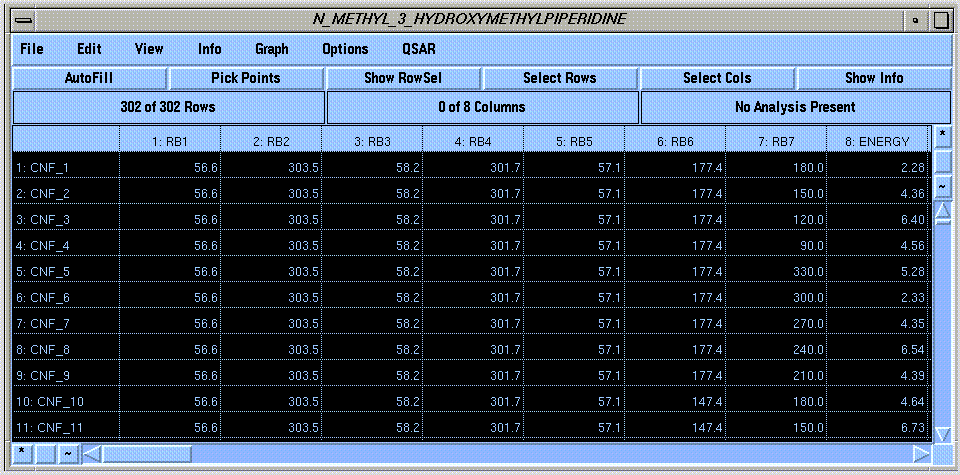
Graphical Visualization of Computed Conformations
Any or all of the conformations stored in a spreadsheet can be graphically displayed using the "Put Rows into Molecular Areas..." command of the spreadsheet's "File" menu:

Notice that the conformations are all superimposed about the anchor atom:

Sorting Conformations by Energy
Conformations in a spreadsheet can be automatically sorted using the "Sort..." command of the "View" menu:
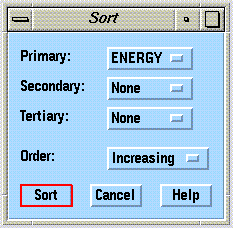
Selections are made in the Sort window:

Energy Plots
Energy plotting is mainly suitable for searches that include up to four bond rotations because of the difficulty in visualizing more than three dimensions at a time (energy + two bond rotations). Plotting rotations of more than two bonds requires the remaining bonds to be fixed at specified dihedral angles. In practice, energy plotting is mainly useful for presenting the results of conformational studies rather than the analysis of conformational data. The two most frequently used plotting methods are iso-energy contours (also called level curves) and three-dimemsional mesh surfaces. The spreadsheet "Graph" menu can be used to produce several different types of energy plots:

An example of an iso-energy contour plot is shown in the following drawing: